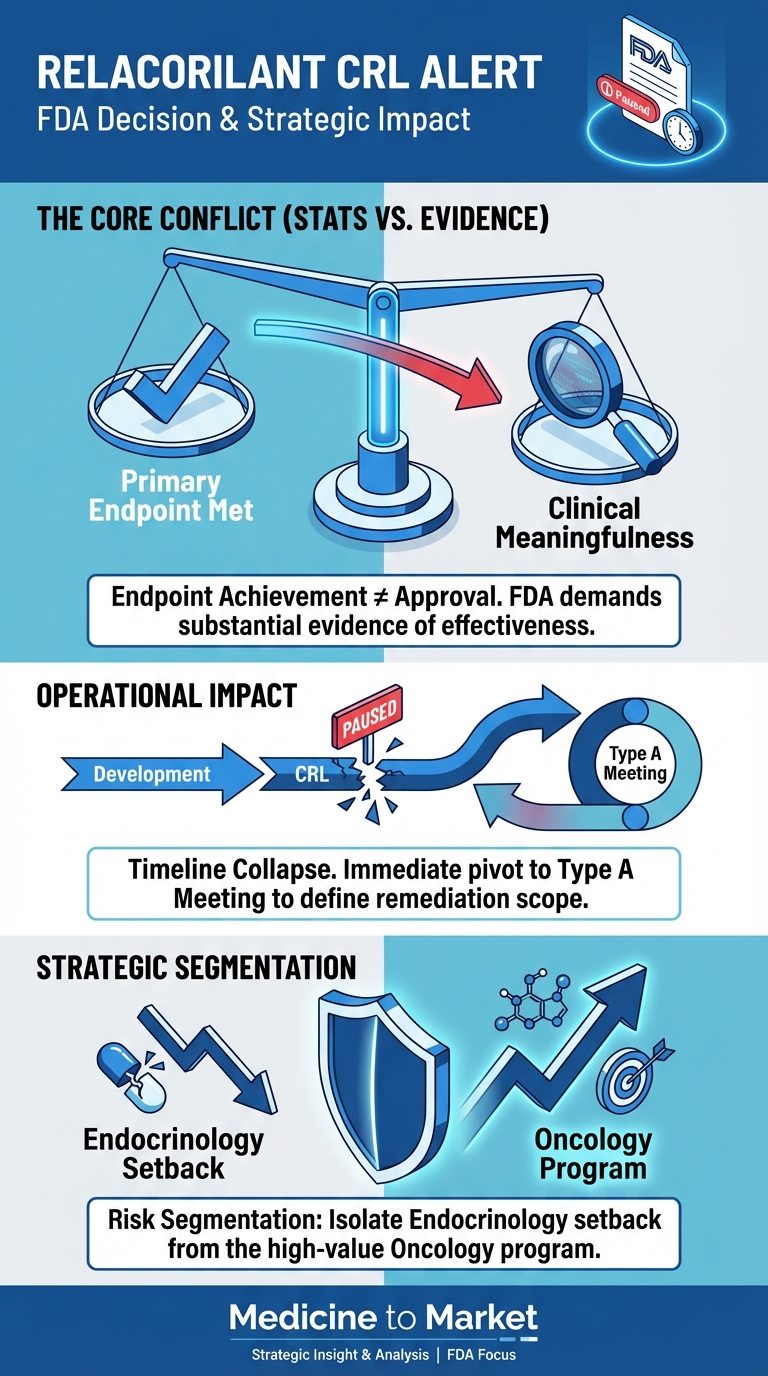
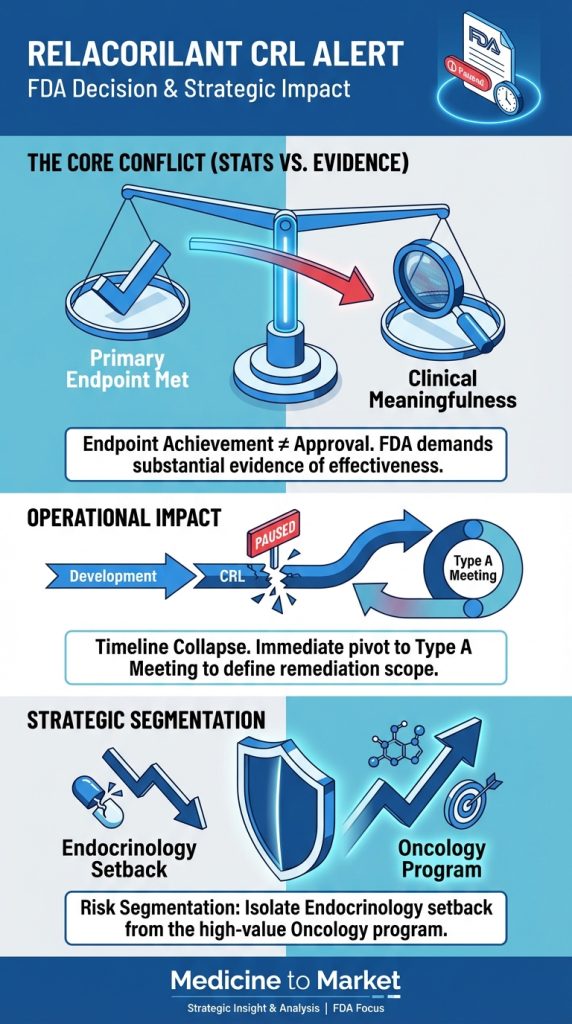
In the evolving landscape of biotechnology and gene therapy, UniQure’s AMT-130 stands out as a beacon of hope for Huntington’s disease, especially as the company gears up to seek FDA approval by
2026.
Recent Phase 2 trial outcomes have shown a striking 75% reduction in disease progression among participants given the highest doses of AMT-130, significantly surpassing initial expectations.
Coupled with a 60% deceleration in Total Functional Capacity decline, these compelling results suggest that UniQure may be on the brink of a breakthrough treatment that addresses the underlying genetic causes of this devastating neurodegenerative disorder.
The implications of this therapy extend beyond its clinical results.
Should the FDA grant approval, it could mark a pivotal moment not only for patients suffering from Huntington’s disease but also for the broader gene therapy sector, which has grappled with a mix of optimism and challenges in recent years.
With the FDA having fast-tracked AMT-130 based on its promise, stakeholders are closely monitoring developments and preparing for discussions around future clinical pathways.

Key Takeaways
- UniQure’s AMT-130 gene therapy shows significant promise, demonstrating a 75% reduction in Huntington’s disease progression.
- The therapy has received FDA fast-track designation, enhancing its chances for timely approval.
- Positive trial results have led to a surge in investor confidence, with the company’s stock tripling following the announcement.
Overview of AMT-130 and Its Development
In a significant development for Huntington’s disease treatment, UniQure is steering towards seeking FDA approval for its innovative gene therapy, AMT-130, by
2026.
Following promising results from a Phase 2 trial, where the therapy showcased a remarkable 75% reduction in disease progression over three years in a cohort of 12 patients at the highest dose, expectations are soaring in the biotech community.
The trial’s results demonstrate a statistically significant advantage when compared to an external control group, solidifying AMT-130’s position as a potential game-changer in this long-challenged therapeutic landscape.
Moreover, additional analyses revealed a 60% slowdown in the decline of Total Functional Capacity, an essential metric for measuring treatment efficacy.
This compelling data has not only fostered optimism but also resulted in a tripling of the company’s stock price, underscoring heightened investor confidence in the therapy’s potential.
As UniQure prepares to engage with the FDA later this year to outline their next steps, the fast-tracking designation by the agency enhances the urgency surrounding AMT-130’s review process.
However, amid this optimism lurks a cautious outlook stemming from uncertainties about the regulator’s approach under current leadership, raising questions about any emerging barriers to final approval.
AMT-130 aims to address the underlying cause of Huntington’s disease by targeting the mutant huntingtin protein notorious for its role in neuronal damage.
Despite previous hurdles in its developmental roadmap and a recognition of the need for operational efficiency, the latest findings have reinvigorated discussions around potential breakthroughs in therapeutic options for this devastating disorder.
Implications of FDA Approval for Huntington’s Disease Treatment
The promising trajectory of AMT-130 reflects not only the potential for significant advancements in Huntington’s disease treatment but also highlights broader implications for the biotech sector as a whole.
Biotech executives must consider how the regulatory landscape, particularly with the FDA’s evolving postures, may influence the timelines and outcomes of future therapies.
If AMT-130 secures approval, it could establish a benchmark for gene therapies targeting monogenic disorders, showcasing a successful transition from genetic research to clinical application.
This case may encourage increased investment in similar therapies, enabling companies to pursue other challenging neurological conditions.
Additionally, the heightened scrutiny on data integrity and trial design after recent industry controversies underscores the necessity for companies to maintain robust, transparent trial protocols as they vie for FDA approval.
Ultimately, AMT-130’s development journey may prompt a reevaluation of genetic therapies within the paradigm of regulatory compliance and market readiness, shaping the strategic priorities of biotech firms looking to innovate in complex therapeutic landscapes.